

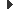
Assaig clínic
Mitjançant la realització d’assajos clínics es pretén l’obtenció sistemàtica i objectiva d’informació. L’objectiu principal dels assajos clínics és l’avenç de la medicina.
Els assajos clínics es defineixen com intervencions experimentals acurades i èticament planificades que resulten de l’aplicació del mètode científic.
En aquesta lliçó es descriuen els diferents tipus d’assajos clínics, els quals s’han classificat atenent diferents criteris. En el segon apartat, l’alumnat profunditzarà en les diferents fases del desenvolupament clínic. A banda, s’especificaran els diferents documents que es generen en un assaig clínic i, per últim, es descriuran les diferents formes de registres dels assajos clínics.
Tipus d’assaig clínic
Recordem que els estudis experimentals són aquells estudis en els quals, com a mínim, una de les variables estudiades es troba sota el control de l’investigador.
En medicina, la investigació de caràcter experimental en la qual participen individus sans o pacients es denomina assaig clínic. Aquests tipus d’estudis constitueixen una investigació meticulosa i curosa. També ha d’estar èticament planificada, ja que es du a terme en éssers humans amb la finalitat de documentar la seguretat, fer més eficaç o trobar el millor tractament per al malalt, o bé, per evitar l’aparició de la malaltia.
Podeu trobar més informació sobre els estudis experimentals, a la lliçó anterior: “Investigació sanitària”.
Atenint-nos a diversos criteris es distingeixen diferents tipus d’assajos clínics:
- Segons el tipus d’intervenció:
- Assaig clínic obert
- Assaig clínic a simple cec
- Assaig clínic a doble cec
- Assaig clínic a triple cec
- Segons el tipus d’aplicació de tractaments i subjectes:
- Assaig clínic creuat (crossover clinical trial)
- Assaig clínic paral·lel (parallel treatment design)
- Assaig clínic factorial
- Assaig clínic seqüencial
- Assaig clínic d’N = 1
- Segons els centres participants:
- Assaig clínic unicèntric
- Assaig clínic multicèntric
Classificació relativa a la intervenció
Tenint en compte el tipus d’intervenció que es fa, trobem diferents tipus d’assaig clínics.
Assaig clínic obert:
Els estudis oberts també es coneixen amb el nom d’estudis no cecs. Quan parlem d’aquests tipus d’estudis, ens referim a aquells assajos en els quals tant el subjecte com l’investigador coneixen a quin tipus de tractament o intervenció mèdica s’ha estat assignat.
S’utilitzen per a la valoració de tècniques quirúrgiques, estudis de deshabituació en drogoaddictes o fumadors, canvis en l’estil de vida, entre d’altres.
Aquest tipus d’estudis són rellevants per dues raons principals:
- Són més fàcils de realitzar que altres tipus d’estudis.
- Confereixen certa comoditat als investigadors a l’hora de prendre decisions.
No obstant això, aquests tipus d’estudis presenten certs inconvenients, sent la possibilitat que apareguin biaixos el més important i/o destacable.
El fet que el participant sàpiga que està rebent un tractament nou pot ser beneficiós des d’un punt de vista psicològic. No obstant això, si l’individu sap que està rebent un tractament estàndard (o no està rebent res si és el cas que no hi ha un tractament estàndard definit), pot reaccionar desfavorablement, sobretot si està al corrent que hi ha altres subjectes “privilegiats” que estan rebent un fàrmac nou. Tot això pot influir no només en la cooperació del pacient en l’estudi, sinó també en la resposta al tractament.
Assaig clínic a simple cec:
En aquest tipus d’assaig clínic el subjecte o pacient ignora quin dels tractaments està rebent, per tant, el subjecte desconeix a quin grup de tractament ha estat assignat. L’investigador, però, sí que ho sap. El que es pretén en aquests tipus d’estudis és neutralitzar l’efecte placebo.
Aquests estudis poden resultar més fàcils pel que fa a la seva organització i monitoratge i, de vegades, són suficients per reduir les possibles desviacions quan es comparen tractaments.
Cal tenir en compte, però, que en determinats assajos cal que l’investigador conegui la intervenció que s’està realitzant per tal de jutjar més adequadament l’evolució del pacient. Un exemple d’utilització freqüent d’aquest mètode on es fa necessari que l’investigador conegui la intervenció és en la valoració del fàrmac per al tractament del càncer. D’aquesta manera es dona la possibilitat de prendre les mesures i accions oportunes.
No obstant això, el problema més gran derivat del fet que l’equip investigador conegui el tractament emprat és la possible influència sobre les decisions preses en el curs de la teràpia, incloent-hi modificació de la dosi, intensitat en la supervisió del pacient, necessitat de teràpia addicional, entre d’altres, la qual cosa podrà influenciar enormement la valoració dels resultats, amb la consegüent inclusió d’una desviació en l’assaig.
Assaig clínic a doble cec:
Quan es fa referència als assajos a doble cec ens referim a aquells estudis en els quals tant el subjecte com l’investigador desconeixen l’assignació als grups de tractament. En aquest tipus d’estudi es tracta de neutralitzar l’efecte placebo i la subjectivitat de l’observador. Són preceptius en estudis d’eficàcia. La seva importància i la seva viabilitat depèn de la malaltia, el tipus de teràpia, el mètode d’avaluació i els recursos disponibles.
El principal avantatge d’aquests assajos és que es redueix el risc de biaixos, considerant-se a priori els estudis preferibles a l’hora d’un millor disseny i garantia d’objectivitat de les dades obtingudes.
Assaig clínic a triple cec:
Quan es parla d’assaig clínic a triple cec ens referim a aquell tipus d’estudi en el qual ni el participant, ni l’investigador observador ni l’investigador que analitza les dades obtingudes coneixen l’assignació ni l’ordre dels tractaments administrats.
Aquests tipus d’assajos clínics es realitzen quan les variables clíniques examinades poden ser interpretades de formes diferents.
La seva finalitat és neutralitzar l’efecte placebo i la subjectivitat de l’observador i de l’avaluador, ja que aquest últim es limitarà a valorar els resultats, sense saber a quin dels tractaments comparats es refereixen.
Classificació relativa a l’aplicació de tractaments i subjectes
Tenint en compte el tipus de tractament a aplicar o els individus que es sotmetran a l’estudi els assajos es poden classificar en:
Assaig clínic creuat (crossover clinical trial):
És l’assaig clínic en el qual ambdós tractaments d’estudi (experimental i control) són administrats a cadascun dels pacients en períodes successius que han estat predeterminats, generalment de manera aleatòria, permeten a cada individu ser el seu propi control (vegeu la figura). Aquest tipus d’estudis garanteix que els individus dels diferents grups tinguin les mateixes característiques, ja que són els mateixos.
En els assajos clínics creuats és necessari que l’administració dels tractaments estigui separada per un període de blanqueig (amb placebo o sense cap tractament), és a dir, un període necessari per eliminar possibles efectes que quedin del que l’individu hagi pogut prendre mentre formava part de l’altre grup.
Aquest període tindrà una durada variable en funció de la patologia en estudi i del temps de vida mitjana d’eliminació de l’agent farmacològic. Aquests períodes controlen el possible cúmul (efecte carry-over) davant de segones o terceres sessions de tractament o preveuen alguns biaixos derivats de partir de condicions clíniques desiguals.
Assaig clínic paral·lel (parallel treatment design):
Els participants són voluntaris. Se seleccionen per tenir l’objectiu d’estudi, és a dir, per exemple tenen hipertensió, colesterol, o qualsevol altra característica.
Als participants se’ls assigna un grup (intervenció o control) de manera aleatòria. L’individu no ha de saber quin grup li ha estat assignat, ja que si ho sabés, podria modificar la seva conducta (vegeu la figura). Després, obtindrem dues respostes:
- Si hi ha resposta estadística significativa (gran diferència), se li donarà una gran importància.
- Si no hi ha gran diferència, no es donarà gaire importància a les conclusions.
Factor de confusió
Es tracta d’un factor que pot tenir l’individu i que pot ser el causant de la millora de la malaltia i, per tant, pot fer que les conclusions siguin errònies. Per exemple: es vol trobar un fàrmac per disminuir els nivells d’hipertensió arterial. Els factors de confusió que poden intervenir serien: l’edat de l’individu, que practiqués esport, consum d’aliments a la dieta que disminueixen la hipertensió arterial, etc. Per tal de corregir aquest factor s’utilitza l’atzar. L’atzar pot fallar en la distribució, per això s’ha de comprovar durant l’estudi.

Assaig clínic factorial: és un tipus d’assaig clínic que permet avaluar diferents intervencions (dosis, fàrmacs, etc.) en un mateix assaig.
Amb una única mostra de pacients s’avaluen al mateix temps per exemple dos tractaments diferents.
Assaig clínic seqüencial: en aquest tipus d’assaig, a mesura que el tractament va avançant, s’estudia el nombre de participants i les mesures que s’han de prendre.
Assaig clínic d’N = 1: són assaigs clínics en què de forma aleatòria s’apliquen dos o més tractaments a un sol malalt. És com un assaig clínic creuat però amb un sol individu. Normalment, es porten a terme amb l’objectiu d’escollir la teràpia (fàrmac i/o dosi) més adequada en un sol malalt.
Classificació relativa als centres participants
Els assajos clínics es poden classificar segons el nombre de centres que participen en l’assaig. Hi ha:
- Assaig clínic unicèntric: s’entén per assaig clínic unicèntric aquell estudi que es du a terme amb pacients o individus sans inclosos a partir d’un únic centre o hospital.
- Assaig clínic multicèntric: es descriuen com assajos clínics multicèntrics aquells estudis en els quals dues o més entitats (hospitals, unitats d’investigació, clíniques o altres) participen en la realització experimental.
Aquests tipus d’estudis es duen a terme sota les directrius d’un mateix protocol i d’un centre coordinador, responsable de la supervisió general de l’assaig clínic. És un coordinador qui unifica les dades.
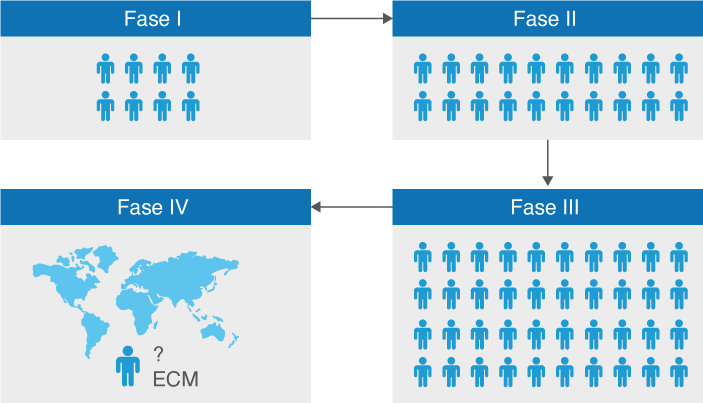
Fases d’un assaig clínic
Abans d’endinsar-nos en les fases pròpies dels assajos clínics, cal esmentar als estudis preclínics, ja que són els antecessors a l’avaluació dels medicaments en éssers humans.
Els estudis preclínics són estudis que es duen a terme sobre cultius cel·lulars i animals en un context científicament controlat.
La finalitat d’aquests estudis preclínics és determinar els possibles riscos per a la salut.
Quan aquests estudis preveuen que el producte candidat a medicament és segur, es poden iniciar les quatre fases en el desenvolupament d’un nou medicament.
ECM
ECM són les sigles d’Ensayos Clínicos con medicamentos. Des del portal web de l’AEMPS, es facilita el tràmit als promotors per tal de realitzar nous assajos clínics: bit.ly/3lQk4Ji.
Les fases d’un assaig clínic són quatre (vegeu la figura). La fase III seria la que representa al grup més gran de persones, mentre que a la Fase IV el món està en ECM (‘assajos clínics amb medicaments’), perquè és quan el medicament ja es comercialitza; tanmateix, ha de passar el temps per veure si hi ha efectes adversos a posteriori (d’aquí l’interrogant).
Assaig clínic en fase I
Aquesta primera fase constitueix el primer pas en la investigació d’un nou medicament en éssers humans i, per tant, es basa en l’administració d’un fàrmac en investigació en humans. S’administra, normalment, en un baix nombre de voluntaris sans, aproximadament en 100 persones, que no presenten cap mena de risc. Donat que els medicaments no seran estudiats en malalts, sinó en pacients sans, en aquesta fase no es podran observar els seus efectes directes.
La fase I d’un assaig clínic vol donar resposta a la pregunta: És segur?
Aquesta primera fase té, principalment, els següents objectius:
- Avaluar la seguretat i la tolerància.
- Conèixer la farmacodinàmica i la farmacocinètica.
- Determinar la dosi màxima tolerada.
- Identificar la millor manera d’administració.
Cal destacar, però, que quan es tracta d’assajos clínics en fase I en oncologia, els participants no són voluntaris sans, sinó pacients sense alternatives terapèutiques.
Quan es tracta d’aquesta patologia, l’objectiu dels assajos clínics en aquesta fase és descobrir i/o determinar la toxicitat limitant de dosi i arribar a una dosi recomanada per als estudis de fase II.
En aquesta primera fase dels assajos clínics amb medicaments, els voluntaris poden percebre una compensació econòmica per la seva participació, tot i que no es tracta d’una compensació prou elevada per poder considerar a aquests participants voluntaris professionals.
Les fases II, III i IV són les d’investigació clínica en malalts.
Assaig clínic en fase II
Aquesta segona fase comprèn la investigació clínica inicial de l’efecte del tractament. Els participants seran voluntaris portadors de la patologia d’estudi que es pretén curar i/o alleugerar. Cal destacar que el nombre de participants tampoc serà molt elevat, ja que serà un nombre aproximat de 200 persones.
La fase II d’un assaig clínic vol donar resposta a la pregunta: Funciona?
Els objectius d’aquesta segona fase són els que s’exposen a continuació:
- Primera avaluació de l’eficàcia del producte.
- Ampliació dels coneixements de seguretat, és a dir, es pretén determinar la incidència d’efectes adversos.
- Farmacodinàmica i farmacocinètica.
- Avaluació de la dosi-resposta per tal de trobar la dosi que serà provada en els assajos en fase III.
Aquesta fase és més perllongada que la fase I.
Aquests estudis poden servir com un procés de selecció per escollir aquells fàrmacs amb potencial real per a ser desenvolupats en fases posteriors (fase III), i complimentar les dades de seguretat en la fase anterior (fase I). Ha de tractar-se d’assajos controlats i amb assignació aleatòria.
Assaig clínic en fase III
En aquesta fase el que es pretén és comparar el fàrmac en investigació amb els tractaments estàndards establerts, sigui placebo o teràpies reconegudes.
Es du a terme tot un procés en el qual es pretén simular la utilització del fàrmac en les mateixes condicions en què es trobaria en el mercat. En aquesta tercera fase participen un gran nombre d’investigadors, ja que es basa a determinar les condicions d’eficàcia i garantia del medicament.
Els participants hauran de patir la malaltia en estudi, i el nombre pot variar d’uns centenars a uns milers de persones.
La fase III d’un assaig clínic vol donar resposta a la pregunta: És més eficaç que el tractament estàndard?
Els objectius d’aquesta fase són els que s’exposen a continuació:
- Confirmar l’eficàcia del principi actiu per la patologia concreta.
- Definir la dosis.
- Definir la pauta de tractament més adequada.
- Avaluar la seguretat del tractament a llarg termini.
Les autoritats sanitàries acostumen a requerir la demostració d’eficàcia en dos assajos clínics de fase III per a permetre la comercialització d’un nou medicament.
Assaig clínic de fase IV
Aquests assajos clínics es realitzen un cop els medicaments ja han estat comercialitzats, i es duen a terme en condicions d’ús autoritzades. Això és degut al fet que, a llarg termini, es desconeix si, tot i haver dut a terme un seguit de controls exhaustius, pot haver-hi un grup de persones a les quals aquests medicaments afecti negativament.
El nombre de participants en aquesta fase és molt elevat, habitualment més de 5000 persones, tot i que es pot arribar a algunes desenes de milers.
La fase IV d’un assaig clínic vol donar resposta a la pregunta: Té altres usos o beneficis?
Els objectius d’aquesta fase són els que es presenten a continuació:
- Avaluació de noves indicacions.
- Determinació de noves vies d’administració.
- Eficàcia i seguretat en determinades condicions d’ús o comparades davant d’altres teràpies ja existents.
- Efectivitat en relació amb el cost.
- Farmacovigilància.
La diferència amb els assajos de fase III és que les condicions d’ús per a la teràpia experimental i la teràpia de control són aprovades en la fitxa tècnica.
Informació i documents generats en un assaig clínic
Un cop tenim clar què és un assaig clínic, els diferents tipus d’assajos clínics que hi ha i les fases per les quals han de passar els medicaments abans de poder comercialitzar-se, és important conèixer les funcions dels responsables dels assajos clínics i tota la documentació que han de preparar per ser avaluats i autoritzats per tal de poder dur a terme els assajos clínics.
Qui participa en un assaig clínic amb medicaments?
Al Reial decret 1090/2015, de 4 de desembre, pel qual es regulen els assajos clínics amb medicaments, els Comitès d’Ètica de la Investigació amb medicaments i el Registre Espanyol d’Estudis Clínics, es concreten les tasques, responsabilitats i competències d’aquelles persones implicades en els assajos clínics (capítol IX). Que són:
- El promotor,
- l’Organització d’Investigació per Contracte (CRO),
- l’investigador,
- el monitor i
- el subjecte d’estudi.
Promotor
El promotor d’un assaig clínic és aquella persona, empresa, institució o organització, responsable de l’inici, gestió i/o finançament de l’assaig clínic.
El promotor pot coincidir amb l’investigador principal. Si aquest fos el cas, aquesta figura hauria d’assumir les responsabilitats de tots dos càrrecs.
Algunes de les funcions del promotor són, entre d’altres, les que s’exposen a continuació:
- Impulsar la investigació.
- Signar, juntament amb l’investigador, el protocol i qualsevol de les seves modificacions.
- Seleccionar a l’investigador.
- Proporcionar la informació bàsica i clínica del producte d’investigació o actualitzar-la al llarg de l’assaig clínic.
- Sol·licitar el dictamen del CEIm i l’autorització de l’Agència Espanyola de Medicaments i Productes Sanitaris.
- Comunicar a les autoritats competents l’inici de l’assaig clínic, així com tota la informació necessària relativa a l’assaig.
- Subministrar de forma gratuïta els medicaments en investigació, garantir que s’han complert les normes correctes de fabricació i que les mostres estan adequadament envasades i etiquetades.
- Responsabilitzar-se de la conservació de les mostres i els seus productes de fabricació i control.
Organització d’Investigació per Contracte (CRO)
L’Organització d’Investigació per Contracte (CRO) és una persona o organització (comercial, acadèmica o altres) contractada pel promotor per realitzar una o més de les funcions o deures del promotor en relació amb l’assaig clínic.
La CRO actua de pont entre el promotor i la resta d’actors que intervenen en la realització de l’estudi clínic (autoritats competents, comitès ètics, centres, hospitals, investigadors, etc.).
Investigador
L’investigador és la persona responsable de la realització de l’assaig clínic en un centre d’investigació, és a dir, és la persona que dirigeix i es responsabilitza de la realització pràctica de l’assaig clínic en un àmbit determinat.
Si és un equip qui realitza l’assaig al centre, l’investigador és el responsable de l’equip, i es pot denominar investigador principal.
L’investigador col·laborador, doncs, és qualsevol membre de l’equip de l’assaig clínic designat i supervisat per l’investigador principal en un centre d’investigació.
Les responsabilitats de l’investigador són les que s’exposen a continuació:
- Signar, juntament amb el promotor, el protocol de l’assaig.
- Conèixer les propietats dels medicaments en investigació.
- Garantir que el consentiment informat és adequat d’acord amb el que s’estableix a la llei corresponent.
- Recollir, registrar i notificar les dades de forma adequada i garantir la veracitat d’aquestes.
- Comunicar/ informar en cas d’esdeveniments adversos o incompliments relatius al protocol establert.
- Garantir la confidencialitat dels participants en l’assaig clínic i la protecció de les dades personals d’aquests.
- Informar periòdicament al CEIm sobre els avanços de l’assaig.
Monitor
El monitor és aquell professional capacitat amb la necessària competència clínica, que s’encarrega del seguiment directe de la realització de l’assaig.
Aquesta figura és escollida pel promotor. Serveix de vincle entre el promotor i l’investigador principal quan aquests no concorrin en la mateixa persona.
El monitor no podrà formar part, en cap cas, de l’equip investigador.
Les responsabilitats del monitor són les que es citen a continuació:
- Treballar d’acord amb els procediments normalitzats de treball del promotor i visitar a l’investigador abans, durant i després de l’assaig (depenent del tipus d’assaig clínic) per tal de comprovar el compliment del protocol.
- Garantir que les dades són enregistrades de forma correcta i completa.
- Assegurar-se que s’ha obtingut el consentiment informat de tots els subjectes abans de la seva inclusió en l’assaig.
- Cerciorar-se que els investigadors i el centre on es realitzarà la investigació són adequats per al propòsit de l’assaig.
- Garantir una comunicació ràpida entre l’investigador i el promotor.
- Comprovar que l’emmagatzematge, distribució, devolució i documentació dels medicaments en investigació és segur i adequat.
- Remetre, al promotor, informes de les visites de monitoratge i de tots els contactes rellevants amb l’investigador.
Subjecte d’estudi
El subjecte d’estudi és la persona que participa en un assaig clínic com a receptor del medicament en investigació o com a control.
Documents generats en un assaig clínic
L’Agència Espanyola de Medicaments i Productes Sanitaris (AEMPS), depenent del Ministeri de Sanitat, Consum i Benestar Social, ha elaborat un escrit en el qual es recull la documentació específica i les instruccions per a la realització d’assajos clínics a Espanya. L’última actualització del document data del 31 de gener de 2022.
Documentació part I: correspon a aquella documentació que s’ha de lliurar conjuntament al Comitè d’Ètica de la Investigació amb medicaments (CEIm) i a l’Agència Espanyola de Medicaments i Productes Sanitaris (AEMPS).
- Carta de presentació.
- Formulari de sol·licitud.
- Autorització del promotor sol·licitant, si és procendet.
- Protocol.
- Resum del protocol.
- Manual de l’investigador o fitxa tècnica del medicament en investigació.
- Fitxa tècnica o manual de l’investigador dels medicaments auxiliars, és a dir, d’aquells medicaments no investigats.
Documentació part II: es tracta d’aquella documentació que només ha de ser lliurada al CEIm.
- Documents en relació amb els procediments i material utilitzats per al reclutament dels subjectes.
- Documents d’informació al pacient
- Consentiment informat.
- Documents d’idoneïtat d’investigadors i dels seus col·laboradors.
- Documents d’idoneïtat de les instal·lacions.
- Quantitats i mètodes de remuneració o indemnització als investigadors i als subjectes participants en l’assaig clínic.
- Pòlissa d’assegurança.
- Assumpció de responsabilitat en absència d’assegurança.
- Compromís de l’investigador.
- Aspectes rellevants del contracte entre promotor i el centre (si s’escau).
- Decisió sobre el PIP (Pla d’Investigació Pediàtrica).
- Assessorament científic emès per una autoritat competent (si s’escau).
A continuació es desenvolupen alguns dels documents citats anteriorment:
Protocol. Les característiques d’un assaig clínic han d’estar íntegrament definides en un protocol. A més a més, l’assaig s’ha d’ajustar al protocol establert. Es tracta, doncs, d’un document on es descriuen els objectius, el disseny, la metodologia, les consideracions estadístiques i l’organització d’un assaig clínic. Generalment, el protocol també proporciona els antecedents i la justificació de l’assaig.
Els apartats que s’han de desenvolupar en el protocol d’un assaig clínic són els que se citen a continuació:
- Resum.
- Índex.
- Informació general.
- Justificació i objectius.
- Tipus d’assaig clínic i disseny.
- Selecció dels participants.
- Descripció del tractament.
- Desenvolupament de l’assaig i avaluació de la resposta.
- Esdeveniments adversos.
- Aspectes ètics.
- Consideracions pràctiques.
- Anàlisi estadística.
Manual de l’investigador. Resum per a l’investigador clínic de les principals característiques del fàrmac a assajar (seguretat i eficàcia).
- Nom genèric, denominació internacional, fórmula química, descripció qualitativa i quantitativa de la composició del fàrmac.
- Estudis preliminars de farmacovigilància animal, farmacodinàmica i farmacocinètica, toxicitat, teratogènesi, carcinogènesi.
- Relació bibliogràfica adequada.
Fitxa tècnica: es tracta del document autoritzat per l’AEMPS en el qual queden reflectides les condicions d’ús autoritzades per al medicament (indicacions, posologia, precaucions, contraindicacions, reaccions adverses, utilització en condicions especials). A banda, recull la informació científica essencial per als metges i altres professionals sanitaris (resum de les dades clíniques, propietats farmacològiques o dades preclíniques sobre seguretat).
Quadern de recollida de dades: es tracta d’un document on es recullen totes les dades relatives als subjectes d’estudi, resultats i observacions obtingudes durant l’assaig clínic. Aquest document s’elabora seguint els requisits establerts en el protocol.
Consentiment informat: el consentiment informat en la investigació biomèdica és un procés de comunicació entre l’investigador i el pacient o voluntari al qual se li proposa la possibilitat de participar en un estudi d’investigació.
El consentiment informat és l’aplicació pràctica del principi d’autonomia o principi de respecte per la persona, és a dir, la capacitat de la persona per prendre decisions segons els seus valors. L’objectiu d’aquest document és brindar informació al pacient perquè la seva decisió sigui lliure.
Registre d’assajos clínics
Amb l’objectiu de crear una font d’informació primària en matèria d’estudis clínics amb medicaments, es va crear el Registre Espanyol d’estudis clínics (REec).
El Registre Espanyol d’estudis clínics (REec) és una base de dades pública, d’ús lliure i gratuït per tots els usuaris, i s’hi pot accedir des de la pàgina web de l’Agència Espanyola de Medicaments i Productes Sanitaris (AEMPS), que és l’entitat que manté i custodia aquesta base de dades.
A l’article 62.1 del Reial decret legislatiu 1/2015, de 24 de juliol, pel qual s’aprova el text refós de la Llei de garanties i ús racional dels medicaments i els productes sanitaris, es dictamina la creació d’un registre nacional d’assaigs clínics. Així, als articles 47 i 48 del Reial decret 1090/2015, de 4 de desembre, pel qual es regulen els assaigs clínics amb medicaments, els Comitès d’Ètica de la Investigació amb medicaments i el Registre Espanyol d’Estudis Clínics, es desenvolupen les característiques i el procediment de publicació del REec.
REec
Podeu accedir al Registro Español de Estudios clínicos en el següent enllaç: bit.ly/2Lec6qn.
Al REec es poden trobar tots els assaigs clínics amb medicaments autoritzats a Espanya des de l’1 de gener de 2013, així com tota la informació respectiva a aquests. A més, des del 2021, s’inclouen, també, tots els estudis observacionals amb medicaments.
Es recull la següent informació:
- De caràcter obligatori, els assaigs clínics amb medicaments d’ús humà que siguin autoritzats per l’Agència Espanyola de Medicaments i Productes Sanitaris.
- De caràcter obligatori, els estudis postautorització de tipus observacional que es realitzaran i hagin estat classificats per l’Agència Espanyola de Medicaments i Productes Sanitaris.
- De caràcter voluntari, altres tipus d’estudis clínics promoguts per entitats públiques o privades, nacionals o internacionals, sempre que tinguin almenys un centre participant radicat a Espanya que inclogui casos o, fins i tot no incloent-hi casos, tingui una contribució espanyola que es consideri significativa.
L’Organització Mundial de la Salut (OMS), en la seva plataforma internacional de registres d’assaigs clínics, defineix uns estàndards per a homogeneïtzar els diferents registres, als quals el REec s’ajusta. Així, al REec es pot trobar informació similar a la que es troba a altres registres d’assaigs clínics:
- Dades generals del formulari de sol·licitud.
- Dates sobre l’avenç de l’assaig que són enviades a l’AEMPS per part del promotor de l’assaig.
- Resum de la justificació de l’assaig en llenguatge llec.
- Informació sobre els centres participants.
- Informació sobre el seu estat en relació amb el reclutament de participants.
Tal com s’ha esmentat al començament d’aquest apartat, el REec té per objectiu ser una font d’informació rellevant per a qualsevol persona interessada en els estudis clínics amb medicaments, ja siguin pacients, professionals sanitaris, investigadors, hospitals, entitats gestores de la investigació, administracions públiques, etcètera. És clau que, per tal de poder complir amb aquesta comesa, és imprescindible que els promotors d’aquests assaigs actualitzin les dades dels seus estudis. Per tal de facilitar aquesta tasca i incentivar a mantenir la base de dades actualitzada, el REec es va actualitzant per fer aquests processos més simples i senzills.
L’existència i el manteniment del REec produeixen una sèrie de beneficis a la societat, aquests són:
- Garanteix que les decisions relacionades amb la salut i cures mèdiques es prenguin amb l’aval de dades científiques públiques i, per tant, reconeguts.
- Garanteix que es posin a disposició de la societat dades i resultats tant positius com negatius dels estudis clínics realitzats.
- Permet que els subjectes participants en tals estudis tinguin informació prèvia de qualitat.
- Evita estudis repetitius o no acceptables, especialment en nens, ancians i altres poblacions vulnerables, potencialment desfavorides o amb dificultats per poder prendre una decisió per si mateixos.
- Detecta aspectes científics poc investigats i ajuda a cobrir aquestes carències.
- Facilita la participació en estudis clínics recentment autoritzats o en marxa i poder, així, aconseguir resultats fiables.
A l’hora de publicar un estudi al REec, cal seguir una sèrie d’orientacions definides a l’article 48 del Reial decret 1090/2015, són:
- Com que el propòsit del REec és proporcionar informació fiable i comprensible per al ciutadà, la informació continguda en alguns camps pot estar limitada quant a extensió i haurà d’estar redactada en un llenguatge senzill i accessible al ciutadà sense especials coneixements científics.
- Per tal que el REec sigui inclòs en la Plataforma Internacional de Registres d’assaigs clínics de l’OMS, la informació haurà d’estar disponible en castellà i en anglès.
- El promotor serà el responsable de la qualitat, l’exactitud i l’actualització periòdica de tota la informació de l’estudi que s’inclogui en el REec. L’Agència Espanyola de Medicaments i Productes Sanitaris verificarà abans de la seva inclusió que les dades són completes i adequades per als seus objectius.
- El registre de l’estudi s’haurà de tramitar, en tot cas, després d’haver obtingut totes les autoritzacions que siguin procedents segons la normativa vigent i abans de la inclusió del primer subjecte en aquest, amb independència que l’estudi hagi estat registrat amb anterioritat a un altre registre públic, inclòs o no a la Plataforma Internacional de Registres d’assaigs clínics de l’OMS.
- La informació dels assaigs clínics amb medicaments, comú amb el Registre d’Assaigs Clínics de la Unió Europea, serà publicada per l’Agència Espanyola de Medicaments i Productes Sanitaris al moment de l’autorització de l’assaig. El promotor d’un assaig clínic amb medicaments disposarà d’un termini màxim de catorze dies naturals a partir de la notificació d’aquesta autorització per completar la informació addicional necessària al REec, amb un resum breu sobre l’assaig i els centres participants.
- Per a la inscripció al REec d’estudis postautorització de tipus observacional, el promotor inclourà les dades de l’estudi en aquell abans del seu inici o en un termini màxim de catorze dies naturals després de l’obtenció de les autoritzacions corresponents segons la normativa vigent per aquells estudis que necessiten autorització prèvia.
- El promotor del tipus d’estudis contemplats a l’article 47.2.c) (estudis d’inscripció voluntària) podrà de forma voluntària optar per la inscripció de les seves dades al REec una vegada que l’estudi tingui un dictamen favorable d’un Comitè Ètic d’Investigació (CEI) i, si escau, l’autorització administrativa corresponent. El dictamen del CEI serà tramès a l’Agència Espanyola de Medicaments i Productes Sanitaris en sol·licitar la inscripció al registre. No obstant això, l’Agència Espanyola de Medicaments i Productes Sanitaris es reserva la potestat última de verificar la informació rebuda i admetre’n la publicació al REeC o, en cas que la informació no pugui ser validada, si no s’inclou.
- L’Agència Espanyola de Medicaments i Productes Sanitaris podrà decidir fer pública una informació parcial sobre un assaig clínic o un estudi postautorització de tipus observacional en què el promotor hagi incomplert els terminis de remissió de la informació, fent constar expressament les dades del promotor així com motius pels quals la informació del registre és parcial. I això amb independència de l’exercici d’aquelles accions sancionadores a què pogués haver-hi lloc, en aplicació de la legislació vigent.
- És responsabilitat del promotor actualitzar periòdicament la informació inclosa en el REec. Aquesta actualització serà obligatòria respecte a la data d’inici de l’estudi a Espanya, els centres participants, la data de finalització del reclutament a Espanya, les modificacions substancials que comportin un canvi en les dades incloses en el registre i la data de finalització de l’estudi a Espanya. L’actualització es farà de manera global i, en tot cas, amb una periodicitat anual fins a la conclusió de l’estudi i la publicació dels resultats.